General Questions |
Illumina Questions |
Oxford Nanopore Technologies (ONT) Questions |
Answers for General Questions
What services does the Next-Gen Sequencing Group provide?
Please see the services page for a detailed list of projects we support. If your project design is not listed, please contact ccrsfhelp@mail.nih.gov or the Next-Gen Sequencing Group director, to discuss the feasibility of a custom project.
Who can order services through the Next-Gen Sequencing Group?
All NIH research labs are eligible to order services through the Next-Gen Sequencing Group. Labs outside of CCR and NIAID will have overhead charges added.
How do I submit a sequencing request?
Please complete a sequencing proposal form at NAS request. There will be an option for Illumina, which you should select for all short read and single-cell projects, and Long Read, which you should select for all PacBio, and ONT projects. You may also contact ccrsfhelp@mail.nih.gov to discuss the available platforms and best choice for your project.
What happens after my sample is submitted?
Before sequencing, we will perform an internal QC to confirm the information in the sample manifest and notify you if any samples do not meet minimum sequencing requirements. You will then be able to choose whether to resubmit those samples or continue and sequence them at your own risks. You will be notified again when the analysis on each sample is completed and available for download.
Answers for General Bioinformatics Questions
What analyses does the NGS Group Bioinformatics group perform?
Currently we offer primary and secondary analyses for all NGS projects, including initial base-calling, demultiplexing, data quality control, and reference genome alignment of NGS reads. We also offer tertiary analyses on a limited basis for collaboration projects, which may include de novo assembly, whole genome small variants and large structural variant analysis, whole genome methylation analysis, long-read full length transcriptomic splice variant detection, and single cell analysis. For all projects, we ensure that every sequence run we deliver meets our high standard for yield, base-call quality, and base alignment percentage and application specific standard metrics that we established.
Sequencing depth and experimental design questions?
Coverage requirements vary by application, library protocol, sequencing platform, and project specific considerations. To provide the best approach for your project, a meeting is setup between you and representatives from our Next-Gen Sequencing Group in order to make recommendations in sequencing platform, library protocol, and other needs.
For assistance in planning your experiment or to discuss specifics of your project please contact Bao Tran (tranb2@mail.nih.gov) or ccrsfhelp@mail.nih.gov.
For bioinformatics consultation please contact Yongmei Zhao (yongmei.zhao@nih.gov) or our group email CCRSF_IFX@nih.gov.
What is required to assure timely processing and delivery of my data?
We recommend an initial consultation with the NGS Group Bioinformatics group to discuss data analysis requirements and to establish expectations. It is also important to specify the reference genome version and annotation build for projects. In addition to model organisms, we also perform data analysis using non-model organisms. For non-model organisms sequencing projects, you will need to provide the reference sequences (FASTA file format or weblink).
If you have any questions regarding your preferred data processing options, please contact Yongmei Zhao (yongmei.zhao@nih.gov) or SF Bioinformatics Team via email CCRSF_IFX@nih.gov.
What types of analysis workflows does the NGS Group use to perform analyses?
We currently provide analyses based on sequencing application type. We have designed and implemented in-house data analysis pipelines that integrate platform/vendor specific data analysis tools with open-source software tools. We released our pipelines to public on GITHUB, which is accessible from the following weblink:
Please contact us at via email CCRSF_IFX@nih.gov if you have any questions.
Currently available data analysis pipelines:
- Illumina Sequencing
- Exome-seq
- Whole Genome Sequencing for SNVs, CNVs and SVs
- RNA-Seq
- miRNA-Seq
- Whole genome methyl-seq
- PacBio Long-read Sequencing
- Iso-seq and single cell MAS Iso-seq
- De novo assembly
- Whole genome sequencing for mutations (SNVs) and structural variant (SV) analysis
- DNA base modification
- 16S amplicon
- Oxford Nanopore Sequencing
- Directed RNA or full-length transcript sequencing
- Adaptive sampling for regions of interest analysis or virus integration sites detection
- Single cell full-length transcriptomic sequencing analysis along with SNV analysis
- Whole genome sequencing for mutations (SNVs) and structural variant (SV) and Copy Number Variation (SNV) analysis
- Single Cell Analysis
- Single Cell RNA-seq and CITE-seq
- Single Cell Immune Profiling
- Single Cell Multiome
What types of data formats will I receive from NGS Group?
For projects using the Illumina sequencing platform, a PDF report containing a summary of the sequencing project (i.e., library and sequencing protocols, sequencing result summary, application-based QC metrics, and software details) and an excel file containing the detailed data analysis results. Depending on the application, you will also receive a html QC report file contains detailed QC statistics and plots for analysis workflows included for that specific application. In addition, you will receive the pass-filtered raw sequence reads in FASTQ format and the reference alignment data in BAM format. BAM files contain base-call and quality score information for all pass-filtered reads, as well as alignment information for reads that have mapped to the reference genome. Additional application specific data files were specified in the deliverable data file types.
For projects using the PacBio sequencing platform, the data delivery choice is driven by the specific needs of the project. For example, when circular consensus processing is performed, the raw subreads bam file, run definition xml files, and the consensus reads (CCS) are included in the data delivery package. If alignment and variant calling are performed, the resulting data are provided within BAM and VCF files. There are also files containing the intermediate results of pipeline processing (such as the read-to-cluster mapping for IsoSeq) are sometimes included. Beyond that, we are happy to deliver any of the files produced by our processing upon request. The content of the data delivery package should be discussed at project definition time.
For standard projects, the deliverable data file types are:
- Sequencing FASTQ/FASTA files
- Alignment BAM files or assembly files
- Data QC statistics reports
- Mapping or variant calling statistics
For projects with secondary and application specific analysis, the deliverable data file types are:
- Exome-seq or WGS Structural Variants Discovery:
- Raw FASTQ files
- Alignment BAM files
- SNP/Indel and structural variant call VCF files
- Sturctural variant call BED file
- Variant annotation files
- QC and variant analysis statistics reports
- RNA-Seq:
- Raw FASTQ files
- STAR-2pass alignment BAM files
- Rsem gene and transcript quantification count matrix files
- QC and RNA analysis statistics reports
- Nanopore Iso-seq:
- Raw data: raw POD5 and FASTQ
- QC and statistics reports: MultiQC report, Squanti3 report and Kraken contamination check
- Analysis data: high quality clustered isoforms, full length cDNAs, Squanti3 results including BAM, GTF, classification table as well as html reports.
- Nanopore De novo Assembly:
- Raw data: raw POD5 and FASTQ
- QC and Statistics reports: MultiQC report, Kraken contamination check
- Analysis data: mapped BAM file, CNV, SNV and SV VCF file
- Nanopore Adaptive Sampling:
- Raw data: raw POD5 and FASTQ
- QC and Statistics reports: MultiQC report, Kraken contamination check
- Analysis data: mapped BAM files, SNV and SV VCF file
- Nanopore Single Cell Sequencing:
- Raw data: raw POD5 and FASTQ
- QC and Statistics reports: MultiQC report, Kraken contamination check
- Analysis data: mapped BAM files, transcriptome matrix, gene matrix, tagged BAM file, Squanti3 filtered results including tagged BAM, GTF, classification table as well as html reports
- The count matrix files to run tertiary analysis with Seurat
- Seurat clustering, SingleR annotations in html report
- Nanopore RNA/DNA base modification:
- Raw data: raw POD5 and FASTQ
- QC and Statistics reports: MultiQC report, Kraken contamination check
- Analysis data: mapped BAM file along with a tagged base modification and BED file
- PacBio Iso-seq/MACS-SC Iso-seq:
- Raw data: CCS/HiFi reads BAM or FASTQ
- QC and Statistics reports: MultiQC report, Squanti3 report and kraken contamination check
- Analysis data: high quality clustered isoforms, full length cDNAs, Squanti3 results
including BAM, GTF, classification table as well as html reports - The count matrix files to run tertiary analysis with Seurat
- Seurat clustering, SingleR annotations in html report
- PacBio WGS Sequencing:
- Raw data: CCS/HiFi reads BAM or FASTQ
- QC and Statistics reports: MultiQC report, kraken contamination check
- Analysis data: mapped BAM file, variant call VCF file
- PacBio De novo Assembly:
- Raw data: CCS reads BAM or FASTQ
- QC and Statistics reports: MultiQC report, assembly report and kraken contamination check
- Analysis data: polished contigs
- PacBio Long Amplicon Sequencing:
- Raw data: CCS reads BAM or FASTQ
- QC and Statistics reports: MultiQC report, kraken contamination check
- Analysis data: Clustered long amplicon consensus, Phasing and variant analysis file
- PacBio 16S Amplicon Sequencing:
- Raw data: CCS reads BAM or FASTQ
- QC and Statistics reports: MultiQC report, kraken contamination check
- Analysis data: an amplicon sequence variant (ASV) table, which records the number of times each exact amplicon sequence variant was observed in each sample
- Single Cell RNA:
- Cell Ranger output
- Seurat clustering, SingleR annotations in html report
- Single Cell ATAC:
- Cell Ranger output
- Signac clustering
- Single Cell Multiome:
- Cell Ranger output
- Single Cell Immune Profiling:
- Cell Ranger output
- Single Cell Fixed RNA Profiling:
- Cell Ranger output
- Single Cell CNV:
- Cell Ranger DNA output
- Single Cell PIPseq:
- PIPseeker output
How do I analyze the data?
The SF typically provides primary and secondary analysis for all applications which include delivery of the FASTQ pass-filtered raw read files and alignment BAM files. For tertiary data analysis for RNA-seq or whole genome sequencing, we offer gene quantification counting files, or variant analysis (both SNVs, SVs) VCF files to the customer. Investigators are expected to provide for their own downstream analyses not offered by the SF bioinformatics group. For investigators interested in performing their own bioinformatics in-house, there are several commercial software options from Illumina, PacBio, and third-party vendors. In addition, many open-source NGS software tools are freely available from Biowulf and other online computing sources.
For investigators interested in need of assistance for downstream NGS data analyses, the CCR Collaborative Bioinformatics Resource (CCBR) provides expert bioinformatics data analysis for the Center for Cancer Research at the NCI free of charge. To contact the CCBR, please submit a request through the CCBR Project Submission Form at https://ccbr.ccr.cancer.gov/project-support/.
How large are the delivery files?
Because NGS sequencing is still a rapidly evolving field, this answer changes regularly. Please contact the bioinformatics group (CCRSF_IFX@nih.gov) for current data delivery file size information.
How are the data files delivered?
Please contact Yongmei Zhao (yongmei.zhao@nih.gov) to discuss your options. The original sequence, alignment, and analysis files are available to download through NCI DME system. To access your project data at DME system, please email Yongmei Zhao (yongmei.zhao@nih.gov) or SF Bioinformatics Team via email CCRSF_IFX@nih.gov to get your NIH account linked to DME system. You will need to register an account for each lab member planning to log in. Please follow DME tutorial (https://wiki.nci.nih.gov/display/DMEdoc) to access and download your project data.
If you or your collaborator does not have NIH account, we recommend to register an account at GlobusFTP (https://www.globus.org/) in order to transfer data via the GlobusFTP site. Please see the following tutorial on registering an account and transferring data:
https://helix.nih.gov/Documentation/globus.html
If you have any issues setting up a Globus account or transferring data via the shared endpoint, please contact us via email CCRSF_IFX@nih.gov.
How long is the data made available to download?
The data files located on NCI DME system currently is depending on the data life cycle defined by data policy. It is available online within 5 years after the initial project data generation. For data files uploaded to Globus system, we make data available for up to 2 weeks starting from the date of our data delivery email announcement. It is the responsibility of the investigator laboratory contact, or bioinformatics contact to ensure that they have retrieved their data promptly. To maintain sufficient data storage for upcoming projects, the analysis files are then archived and stored for an additional four weeks for Globus data transfer.
If your data is no longer available for download, please contact the SF bioinformatics group and we can re-run the data processing and alignment as necessary. However, please note that it may take longer to receive the re-analyzed data due to resource conflicts with current production runs. Whenever possible, it is best to download the data in a timely manner after receipt of the delivery notice.
What is the yield per run for different sequencing platforms?
The following table provides the example yields per sequencing instrument types based on applications supported at NGS Group by the vendor supported chemistry and flow cell types. Actual performance parameters may vary based on sample type, sample quality, and clusters passing filter
| Sequencing Platform | Specification Website |
| Illumina NovaSeq Xplus | https://www.illumina.com/systems/sequencing-platforms/novaseq-x-plus/specifications.html |
| Illumina NextSeq 2000 | https://www.illumina.com/systems/sequencing-platforms/nextseq-1000-2000/specifications.html |
| Illumina MiSeq | https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html |
| PacBio Revio System | https://www.pacb.com/wp-content/uploads/Revio-specification-sheet.pdf |
| Oxford Nanopore PromethION | https://nanoporetech.com/products/promethion |
For further questions, please email Yongmei Zhao (yongmei.zhao@nih.gov) or SF Bioinformatics Team via email CCRSF_IFX@nih.gov.
Answers for Illumina Questions
How do I submit samples for Illumina sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Next-Gen Sequencing Group team and the NAS request submitted. Illumina service is listed under Next-Gen Sequencing Group –Illumina (CCR).
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery. Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to Jyoti Shetty prior to sending your samples.
What are the requirements for submitted samples for Illumina sequencing?
Sample Quantity/Quality Requirements and Recommendations:
All samples are shipped in dry ice and the individual (1.5-2 ml) tubes are labeled clearly
| Type of library | Minimum DNA/RNA Requirement for Library Construction | Recommended DNA/RNA for Optimal Library Construction | Maximum Sample Volume Requirement for Library Construction | Additional requirements |
| ChIP DNA Sequencing | 5 ng | 10 ng | 30 µL | Bulk of the DNA fragments in the 100-300 bp range |
| gDNA Sequencing | 100 ng | 1 µg | 30 µL | DNA should be as intact as possible with no contamination, OD260/280 1.8–2.0 |
| mRNA Sequencing | 25 ng | 1 µg | 30 µL | RIN should be at least 8.0, DNase treated |
| mRNA ultralow | 100 pg | 10 ng | 10 µL | RIN should be at least 8.0, DNase treated |
| microRNA Sequencing | 100 ng | 1 µg | 6 µL | |
| Total RNA Sequencing | 10 ng | 1 µg | 10 µL | DNase treated, FFPE and degraded RNA can be used; DV200 < 30% not recommended |
You can use any extraction protocol as long as the DNA/RNA samples meet our sample requirements.
Answers for PacBio Questions
How do I submit samples for PacBio sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Next-Gen Sequencing Group team and the NAS request submitted. PacBio service is listed under Next-Gen Sequencing Group – Long Read Technology (CCR)
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery). Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to caroline.fromont@nih.gov prior to sending your samples.
What are the requirements for submitted samples for PacBio sequencing?
All samples must be sent in a 1.5 ml or 2 ml tubes.
Some requirements might be project dependent such as input of DNA if multiplexing or sequencing on multiple SMRTcells. Please contact us for more details.
Quality and quantity requirements are listed in the table below:
| Type of Library | Minimum DNA/RNA Quantity Requirement | Recommended DNA/RNA Quantity Requirement | Minimum Concentration Requirement | Quality Requirements |
| WGS | 1.5 µg | 5 µg | n/a | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| Amplicons (< 5000 bp) | 200 ng | 500 ng | n/a | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| Amplicons (> 5000 bp) | 300 ng | 800 ng | n/a | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| HLA (Class I) | 250 ng | 1 µg | 20 ng/µL | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| 16S | 2 ng | 10 ng | 500 pg/µL | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| MAS Single Cell | 15 ng | 50 ng | 1 ng/µL | OD260/280:1.8-2.0 OD260/230:1.7-2.2 |
| WTS | 300 ng | 1 µg | 50 ng/µL | RIN ≥ 8.0 |
What happens during PacBio library preparation?
After initial sample QC, we proceed with library preparation. Depending on the project, the samples will be handled differently prior to PacBio library preparation. For amplicons samples, we first perform an AMPure bead clean-up that also allows us to concentrate the samples if necessary. For gDNA samples, we shear the samples to the targeted size depending on the project need and perform an AMPure bead clean-up to concentrate the samples. For WTS, we generate cDNA using polydT primers and TSO allowing us to target full length transcripts with a polyA tail. During PacBio library preparation, fragments undergo damage repair, end-repair/A-tailing and adapter ligation. The adapters are hairpin adapters and, ligated to double stranded DNA, they form a circular molecule necessary for PacBio sequencing. Barcoded hairpin adapters are also available if the project requires pooling of multiple samples. The libraries are then cleaned using AMPure beads and we perform a QC prior to setting up a sequencing run.
What are PacBio HiFi and CCS reads?
CCS stands for Circular Consensus Sequences. CCS are produced for sequencing libraries with insert size shorter than 25 kb. For CCS, the circular template (dsDNA with hairpin adapters) generated during library preparation is read multiple times and produces numerous read passes (subreads). Those subreads are then used to call a consensus sequence and generate highly accurate reads. Four passes of the molecule usually yield Q20 data while 8 passes should yield Q30 data. HiFi reads are CCS reads with > Q20.
For a quick explanation of SMRT sequencing, please watch the following PacBio video https://youtu.be/_lD8JyAbwEo
On the PacBio website: https://www.pacb.com/technology/hifi-sequencing/
What is the estimated output for PacBio sequencing?
Please note that these are estimates only as both library type and insert size are going to influence the output and it is subject to change. The Sequel II is estimated to produce 3-4.5 million raw reads. For RNA iso-seq libraries you can expect to get 3-4 million CCS reads. For WGS libraries, you can expect to get about 20-30 Gb of HiFi reads.
What can I sequence on one SMRT Cell 8M?
According to PacBio, one SMRT cell is enough to sequence a genome up to 2 Gb and a whole transcriptome, detect structural variants in up to 2 samples of ˜3 Gb genome, and multiplex numerous amplicons. For variant detection (single nucleotides, indels and structural variants) in a ˜3 Gb genome, using at least 2 SMRT cells is recommended.
See “What can you do with one SMRT cell?” for more information.
How can I extract HMW DNA?
PacBio has released a list of HMW DNA extraction protocols and QC methods that can be found at DNA preparation technical note
How can I perform target enrichment for PacBio Iso-seq?
If you are interested in long read isoform sequencing but are focused on only one or a few genes you may consider a target enrichment protocol. This protocol relies on hybridization of biotinylated probes to your cDNA target of interest and subsequent pulldown with streptavidin beads. The enriched cDNA is then amplified and prepared for PacBio sequencing. To design probes for your project please contact IDT at NGSDesign@idtdna.com or fill out a probe design request at https://go.idtdna.com/Request-consult-NGS-xGen-Custom-Hyb-Panel. To complete your sequencing request you will need to submit your probe panel in addition to your RNA samples. Please contact us for more details.
Answers for Oxford Nanopore Technologies (ONT) Questions
How do I submit samples for ONT sequencing?
Before submitting samples, ensure that the sequencing project has been discussed with the Next-Gen Sequencing Group team and the NAS request submitted. ONT service is listed under Next-Gen Sequencing Group – Long Read Technology (CCR)
You may then submit samples by delivering them at the ATRF Room D3040 (instructions for sample delivery). Be sure to include a sample manifest form with your submission as well as to send an electronic version of the form to Juanma Caravaca prior to sending your samples.
What happens after my sample is submitted?
Before sequencing, we will perform an internal QC to confirm the information in the sample manifest and notify you if any samples do not meet minimum sequencing requirements. You will then be able to choose whether to resubmit those samples or continue and sequence them at your own risks. You will be notified again when the analysis on each sample is completed and available for download.
What are the requirements for submitted samples for ONT sequencing?
All samples, except ONT Ultralong, must be sent in 1.5 or 2 mL tubes in dry ice. For ONT Ultralong, send the cells as a frozen pellet or cryopreserved vial in dry ice.
Quality and quantity requirements are listed in the table below:
| Type of library | Minimum DNA/RNA Requirement for Library Construction | Recommended DNA/RNA for Optimal Library Construction | Maximum Sample Volume Requirement for Library Construction | Additional requirements |
| WGS | 1 µg | 4 µg | 48 µL | HMW DNA |
| WGS Adaptive Sampling | 2 µg | 5 µg | 48 µL | HMW DNA |
| WGS Ultralong | 6 million human cells or the cell number equivalent to 40 µg of DNA | n/a | n/a | n/a |
| Direct RNA Sequencing | 50 ng of poly(A) tailed or 500 ng total RNA | 150 ng of poly(A) tailed or 1 µg total RNA | 9 µL | RIN ≥ 8.0 |
How can I extract HMW DNA?
We recommend the HMW Circulomics kit (NB-900-001-01) for DNA extraction.
What is the estimated output for ONT sequencing?
- GridION: up to 50 GB per flow cell
- PromethION 2 Solo: up to 200 GB per flow cell
What is sample processing workflow for single cell projects?
Upon receiving single cell suspension, we check for the quality of cells and wash the cells a couple of times with PBS+BSA before loading it onto a microfluidic chip for the capture of single cells in a nanoliter size droplets along with the barcoded beads. RT takes place inside the droplet and then we break the droplets and PCR amplify the cDNA in bulk. We then purify the cDNA and check the quality. Generally, the quality of cDNA correlates very well with the final sequencing results. We then make Illumina compatible libraries from these cDNAs and sequence it on sequencer.
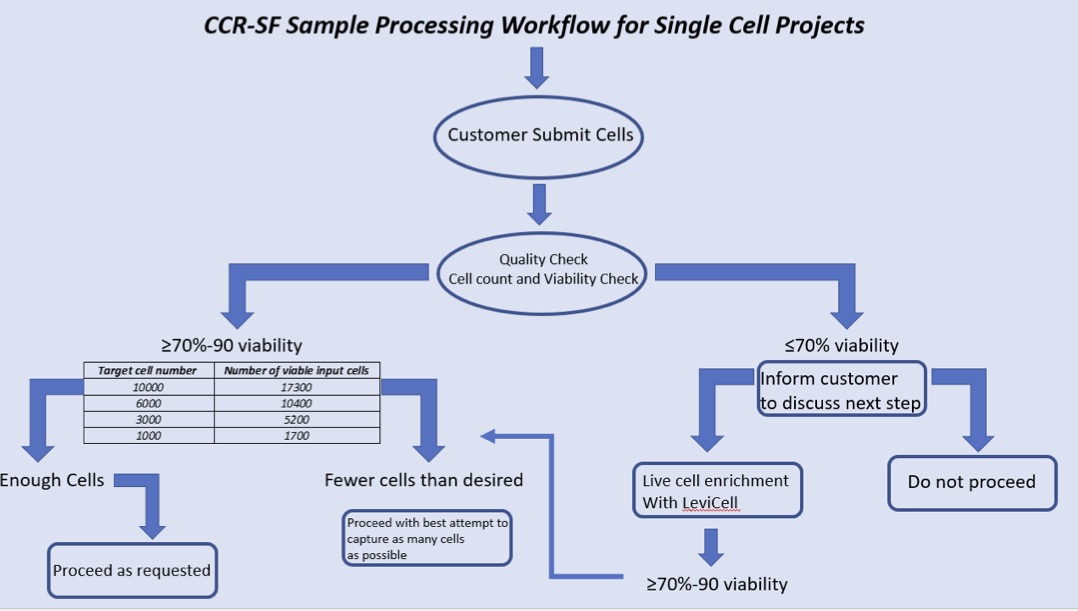
What is Single Cell RNA-Seq?
Single-Cell RNA-Seq provides transcriptional profiling of thousands of individual cells. This level of throughput analysis enables researchers to understand at the single-cell level what genes are expressed, in what quantities, and how they differ across thousands of cells within a heterogeneous sample.
Do dead cells impact the data quality?
10X Genomics Single Cell Protocols require suspensions of viable (90% optimal, 70-90% acceptable), single cells as input. Dead cells easily lyse, resulting in the release of ambient RNA. This cell-free RNA can contribute to the background noise of the assay and will compromise the quality of single cell data.
Clusters of dying cells typically have relatively higher levels of mitochondrial expression, lower gene counts, and more ambiguous cell type identification scores that equally or comparably match multiple major cell types.
How many cells do I need to provide?
We recommend > 1×106 cells/mL – minimum 200,000 cells/mL to load 10K live cells for non-hashing experiments and 20K-30K live cells for cell hashing experiments.
How many cells can I expect to get information for?
The capture rate of 10X is approximately 60%, depending on cell type and cell quality. When you load 10K (70-90% live) cells, you will capture around 6K cells.
How many reads do I need for my experiment?
We aim to provide 20K-50K reads/cell for gene expression libraries, 10K reads/cell for V(D)J enriched samples, 5K-10K reads/cell for CITE-Seq libraries, 50K reads/cell for single cell ATAC libraries, 20K-50K reads/cell for single cell multiome libraries as recommended by 10X genomics.
What 10X applications do you support?
We support following 10X assays: single cell gene expression (3’ and 5’), single cell immune profiling (V(D)J, TCR, BCR), Single cell multiome ATAC+Gene expression, Single Cell ATAC. We also support Mission Bio Tapestri single cell targeted DNA application.
What buffers should I use to resuspend my cells on the day of submission?
10X recommended to use 1X PBS (calcium and magnesium-free) containing 0.04% weight/volume BSA (400 μg/ml) for washing and resuspension. It is also possible to use most cell culture media with up to 10% FBS or up to 2% BSA to maintain cell health with little to no adverse downstream effects. Media should not contain excessive amounts of EDTA (> 0.1mM), or magnesium (> 3mM) as those components will inhibit the reverse transcription reaction. Any surfactants (Tween-20, etc.) should also be avoided as they may interfere with GEM generation.
How should I prepare and send my samples?
We accept fresh and cryopreserved cells. Please use 10X Genomics recommended or supported cryopreservation protocols for your cells, and follow the 10X Genomics full cell preparation guide.
Fresh samples need to be at CCR_SF within 1-1.5 hour after dissociation. Fresh samples need to be loaded onto the 10X machine as soon as possible after dissociation. Please bring your samples before 2pm!
- Cell parameters: Recommended cell number: > 1×106 cells/mL. Minimum 200,000 cells/mL
- Viability: Recommended > 90%. Acceptable 70%-90%.
- Container: Please use 15mL conical Falcon tubes or 2 mL Eppendorf tubes
- Medium: You can hand your dissociated cells over in cell culture medium (up to 10% FBS or up to 2% BSA) or in 1X PBS/0.004% BSA.
- abeling: Please label tubes clearly and use permanent markers or labels. Always label your tubes on the lids and the side. Please use short unambiguous names (e.g., CTRL, IFN1).
- Temperature: Please deliver your fresh cells on ice. Please ship your cryopreserved samples on dry ice.
What is the cost per sample?
All the information about pricing is listed on our website.
What is included in the price?
We provide full service which includes administrative services, consultations, advice on experimental design, 10X genomics reagents, sample QC prior to loading, cDNA QC, post library generation QC, primary bioinformatic analysis (using the Cell Ranger pipeline).
What is the Turn Around Time for your single cell core?
It takes about 6-8 weeks from the time the sample is submitted, to data delivery after running the CellRanger pipeline. Turn around time can increase for large projects (> 48 samples) and close to the end of the fiscal year.
How should I schedule the experiment?
Please email our scientific team. They will reply you and may arrange a short meeting to discuss the project. The scheduling should be at least 2 weeks in advance. When your samples are ready, you will need to submit a NAS request listed under Next-Gen Sequencing Group, Illumina (CCR). Please fill out a sample manifest form and send back to us.
Sequencing Cost Calculator
*For full estimate, including library prep., etc., click "Next".
| Sample_ID | Library Type | Target Cell Number (in thousands) | Target Reads per Cell (in thousands) | Number of Reads per Sample (in millions) | Total Reads (in millions) |
|---|
*For full estimate, including library prep., etc., click "Next".
*For full estimate, including library prep., washing kits, etc., click "Next".
*For full estimate, including library prep., washing kits, etc., click "Next".
| Sample_ID | Library Type | Target Cell Number (in thousands) | Target Reads per Cell (in thousands) | Number of Reads per Sample (in millions) | Total Reads (in millions) |
|---|
*For full estimate, including library prep., qc, etc., click "Next".
| Sample_ID | Library Type | Target Cell Number (in thousands) | Target Reads per Cell (in thousands) | Number of Reads per Sample (in millions) | Total Reads (in millions) |
|---|
*For full estimate, including library prep., qc, etc., click "Next".
Add-On Modalities (Optional)
Add-On Modalities (Optional)
Add-On Modalities (Optional)
| Description | Unit | Quantity | Total | |
|---|---|---|---|---|
| Library Prep | ||||
| Kit | ||||
| QC CostPooling Cost | QCPooling | 1 | ||
| Grand Total: | ||||
|
* You have an option to request a QC run. But we do not guarantee the read distribution if you send us already pooled libraries. |
||||
|
* This is only an estimate, generated on July 3, 2026 at 7:02 am. Please reach out to Bao Tran (tranb2@mail.nih.gov) or Jyoti Shetty (jyoti.shetty@nih.gov) for a more accurate quote. |
||||
| Description | Unit | Quantity | Total | |
|---|---|---|---|---|
| Library Prep | ||||
| Nanopore flow cell | Consumable | |||
| Washing kit | Consumable | |||
| Reloading reagents | Consumable | |||
| Grand Total: | ||||
|
* This is only an estimate, generated on July 3, 2026 at 7:02 am. Please reach out to Bao Tran (tranb2@mail.nih.gov) or Kayla Kline (kayla.eury@nih.gov) for a more accurate quote. |
||||
| Description | Unit | Quantity | Total | |
|---|---|---|---|---|
| Library Prep | ||||
| Revio SMRT Cell | Consumable | |||
| Grand Total: | ||||
|
* This is only an estimate, generated on July 3, 2026 at 7:02 am. Please reach out to Bao Tran (tranb2@mail.nih.gov) or Kayla Kline (kayla.eury@nih.gov) for a more accurate quote. |
||||
| Item | Type | Quantity | Unit Price | Total |
|---|---|---|---|---|
| Grand Total: | ||||
|
* This is only an estimate, generated on July 3, 2026 at 7:02 am. Please reach out to Bao Tran (tranb2@mail.nih.gov) or Lirong Peng (Lirong.peng@nih.gov) for a more accurate quote. |
||||
This calculator functionality is not yet available.